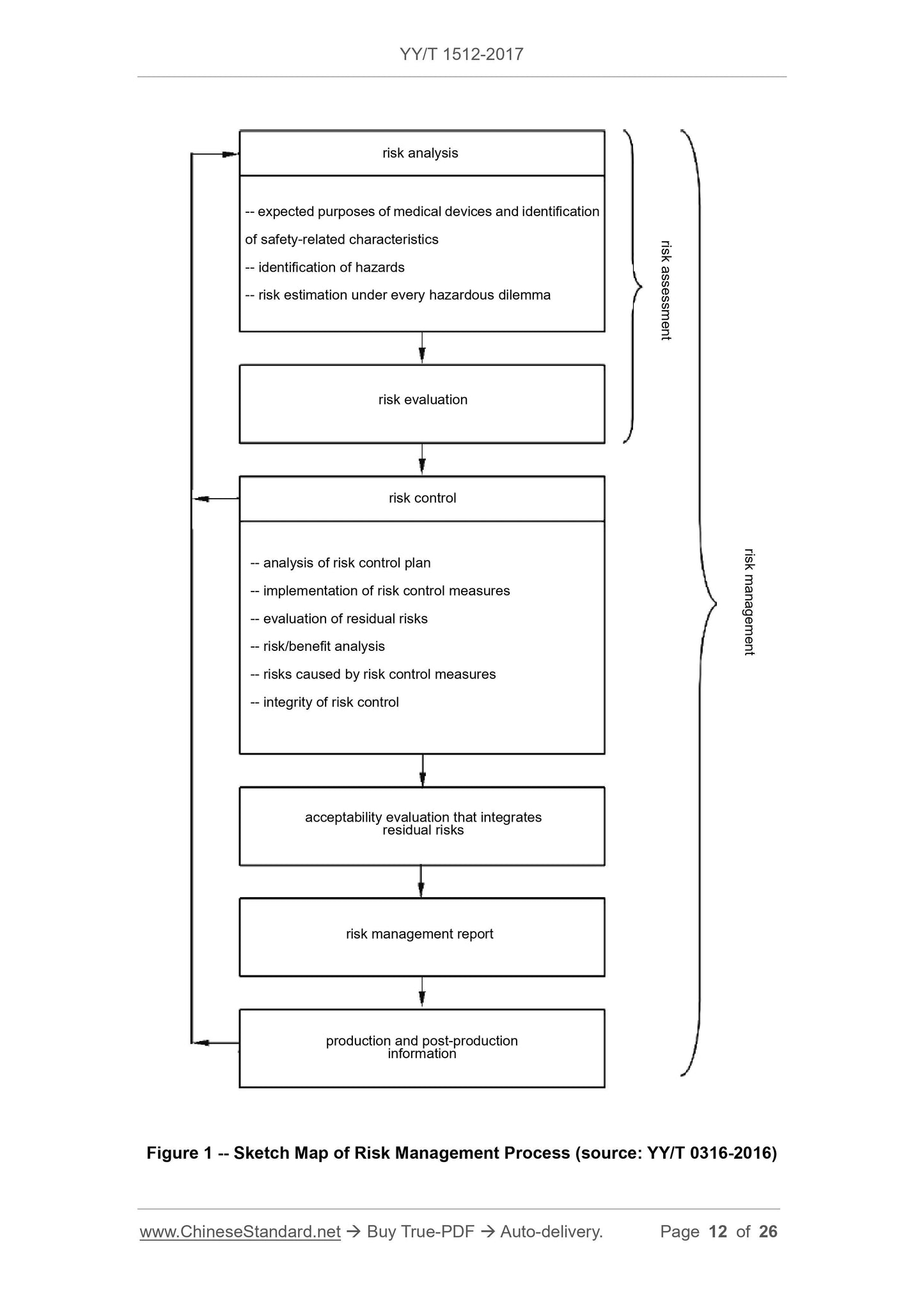
This Standard is applicable to the biological evaluation of medical devices in accordance with the requirements in GB/T 16886.1-2011. This Standard does not add or modify the requirements in GB/T 16886.1-2011. This Standard does not include requirements of regulatory inspection or certification and assessment activities. This Standard is applicable to all the biological evaluation of various types of medical devices, including active, passive, implantable and non-implantable medical devices.
Basic Data
Standard ID
YY/T 1512-2017 (YY/T1512-2017)
Description (Translated English)
Biological evaluation of medical devices - Guidance on the conduct of biological evaluation within a risk management process
Sector / Industry
Medical Device and Pharmaceutical Industry Standard (Recommended)